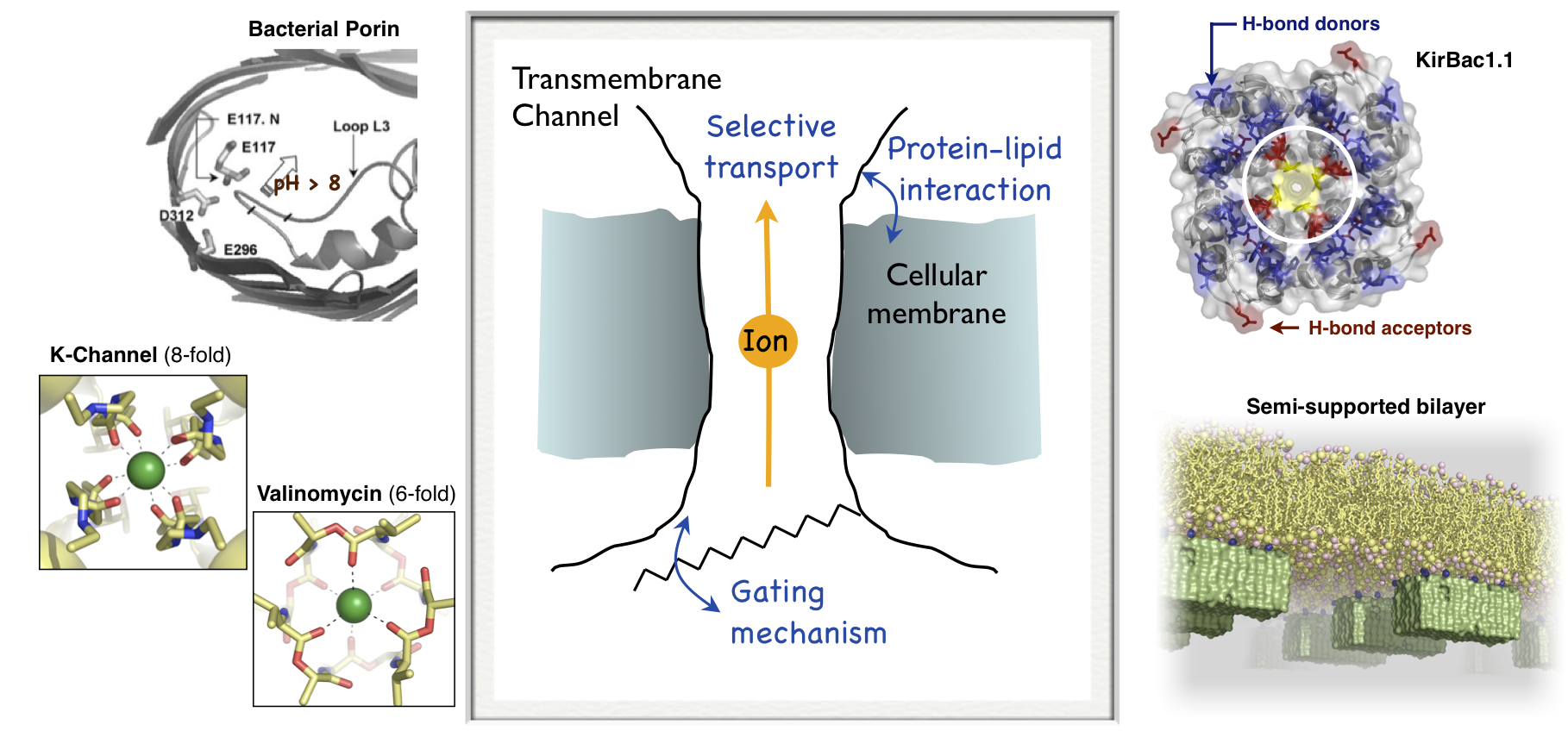
Channels and transporters
Channels and transporters particpate in many physiological processes, including nerve conduction and muscle regulation, by tightly controlling the concentration gradients of ions and solutes across cell membranes. We are using quantum and molecular mechanics simulations to understand the design principles in these proteins that allow them to selectively transport ions and also open-and-close in response to physiological stimuli.
Collaborators: David Rogers,
Thomas Talyor-Clark
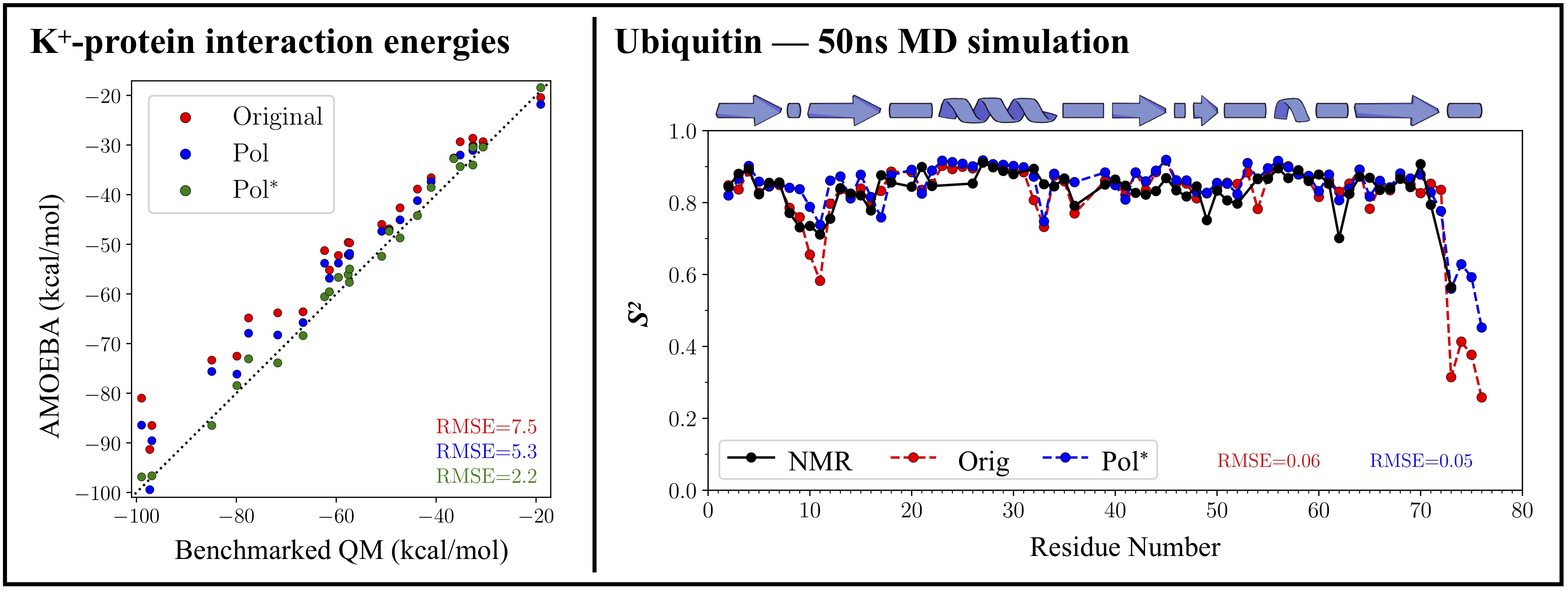
Accurate and efficient modeling of interactions of biomolecules with ions
Ions participate in numerous biochemical pathways by interacting directly with biomolecules and after shedding, at least partially, their solvating water molecules. Consequently, mechanistic insights into these processes require a fundamental understanding of how the energetics/structures/dynamics of ion-binding differ between hydrated and biomolecule-bound states. While first principles quantum mechanical models can yield reliable estimates for relative binding energies, estimates for thermodynamics and ion-binding response are subject to limitations from conformational sampling and system size. In contrast, molecular mechanics models can technically get past sampling/system-size issues, but they suffer severely from accuracy. Polarizable molecular mechanics models do offer a long term sustainable compromise, and they do outperform non-polarizable models, but there is now clear evidence that errors in such models can also exceed 10 kcal/mol. Such errors can, and have led to incorrect predictions/interpretations in mechanistic cause-effect relationships. We are conducting high-level quantum mechanical studies to understand these interactions and identify the essential physics missing in molecular mechanics models. We have also started to plug this physics into the polarizable AMOEBA model (developed by Ponder, Ren and coworkers). Combined with a many-body NB-fix approach, our most recent recalibration of AMOEBA protein force field reduces errors in Na
+-protein, K
+-protein and Mg
2+-protein interaction energies from 8.7 to 2.7 kcal/mol, 9.6 to 2.6 kcal/mol and 17 to 6 kcal/mol, respectively. The recalibration of AMOEBA protein parameters does not affect its intrinsic reliability in predicting protein structure and dynamics in the aqueous phase. The latest release (amoebabio18_hfc2023.prm) also contains new parameters for triphosphates and Mg
2+-phosphate interactions that improve MD simulations of Mg
2+.Protein.Nucleotide complexes.
Collaborators: Sagar Pandit,
Alexandre Tkatchenko,
Peter Nagy
Latest versions of AMOEBA Force Fields:
1)
amoebapro13_hfc22.prm,
2)
amoebabio18_hfc23.prm,
amoebabio18_hfc23.README.txt
Membrane remodulation by extrinsic constraints
The physical properties of lipid bilayers can be remodeled by a variety of environmental factors. Recent advances in nanotechnology, such as those used in nano-biosensing, drug delivery vehicles and synthetic cells, bring to the forefront a new class of extrinsic constraints for remodeling lipid bilayers. In this next-generation technology, membranes are supported over nanoporous substrates -- the nanometer sized pores in the substrate are too small for bilayers to follow the substrate topology, and consequently the bilayers hang over the pores. Experiments demonstrate that nanoporous substrates remodel lipid bilayers differently from continuous substrates. We are using molecular dynamics and self-consistent mean field theory to make sense of these observations from a molecular standpoint and construct predictive models that can be used in future to synthesize designer supported membranes.
Collaborators: Sagar Pandit,
Larry Scott,
Jeffrey Brinker

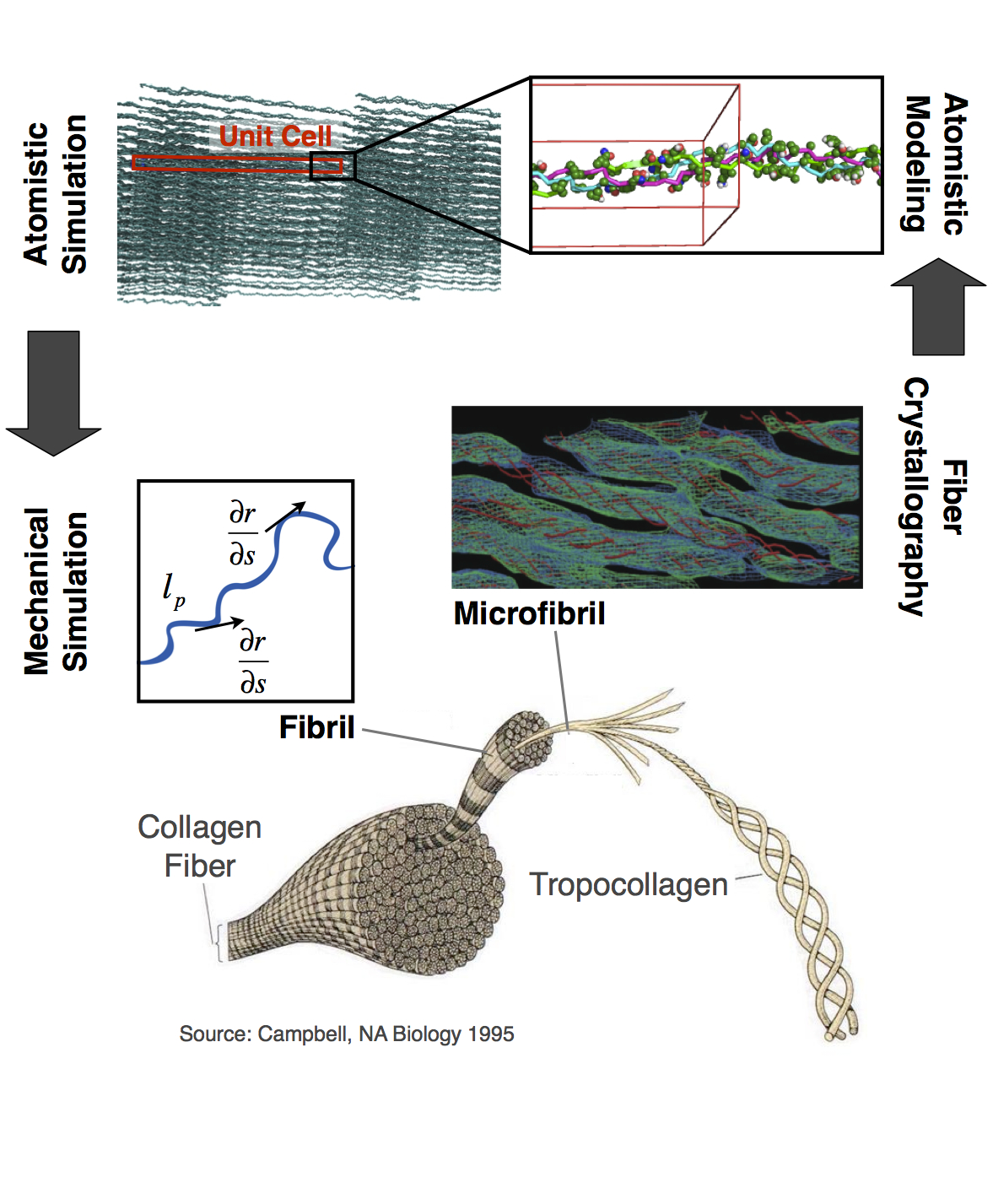
Collagen mechanics and assembly
Collagen is the primary constituent of the extracellular matrix of animal connective tissue. There are many different types of collagen,
and among them, Type I is the predominant collagen in mature tendons and ligaments, where it gives them their load-bearing
mechanical properties. Fibrils of type I collagen are formed by the packing of polypeptide triple helices. Higher-order
structures like fibril bundles and fibers are assembled from fibrils in the presence of other collagenous molecules and non-collagenous
molecules. We are developing and using atomistic and coarse-grained simulation techniques to gain detailed insight into this assembly process and also the dependence of mechanical properties on structural hierarchy. This will be key to understanding the response of various biological factors on collagen, including congenital disease-causing mutations, and will also aid in engineering new collagen-based materials.
Collaborators: Joseph Orgel,
Jay Schieber
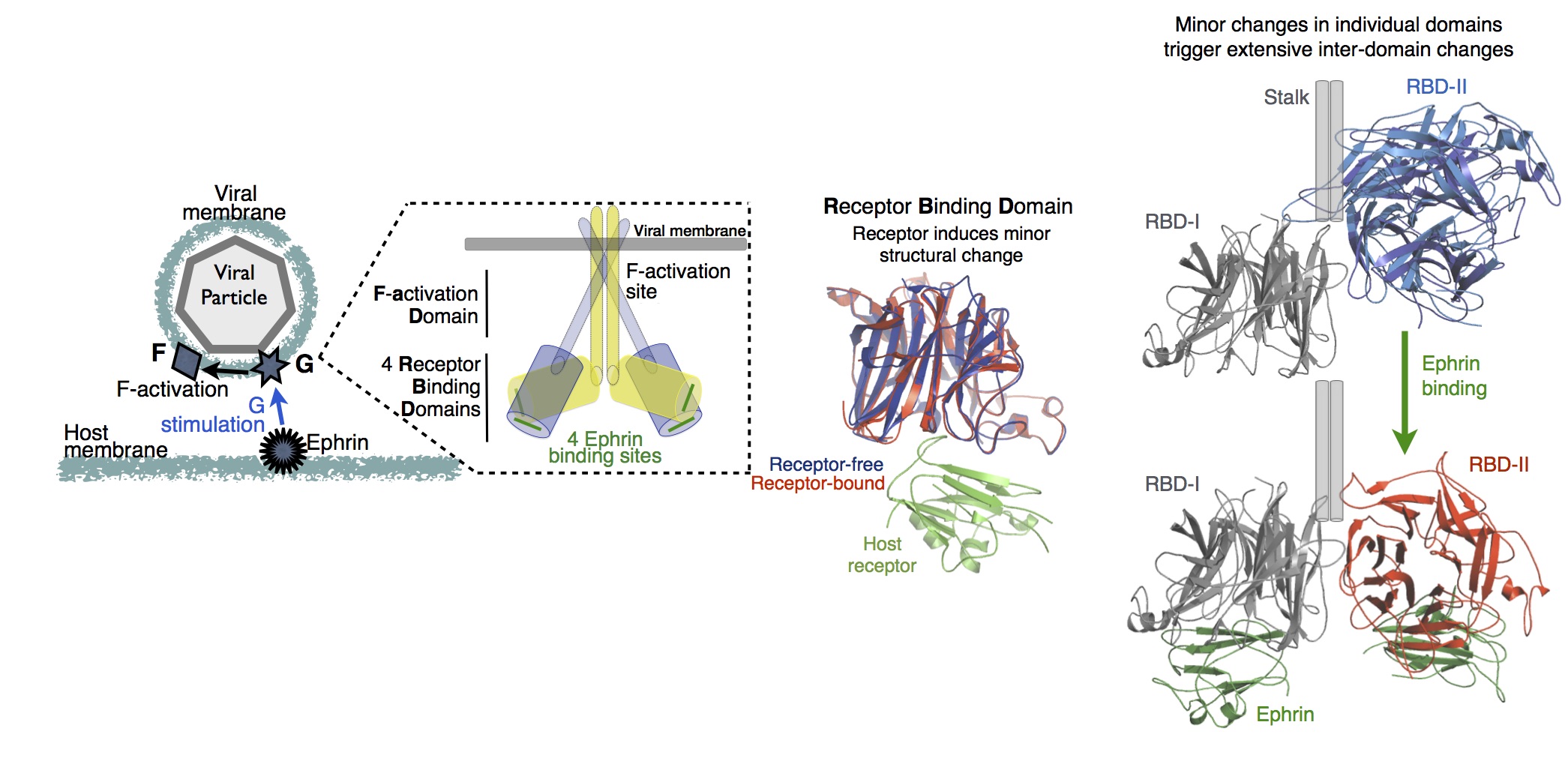
Paramyxovirus entry and dynamic allostery
Paramyxoviruses, such as the Measles Virus, Newcastle Disease Virus and the recently emerged Nipah Virus, are highly virulent
and cause numerous serious diseases in humans and farm animals. Toward developing antiviral therapies that have enhanced resistance to viral mutations,
we are constructing a detailed model of the very first step of their infection process - the allosteric simulation of the viral attachment protein by host cell receptors.
Stimulation of the viral attachment protein by host receptors is essential for the effective fusion of the virus with the host cell.
The primary challenge here is that, just as in many eukaryotic proteins,
including GPCRs, PDZ domains and T-cell receptors, receptor binding induces minor structural changes (less than 0.1 nm)
in the receptor binding domain of the viral attachment protein, and consequently, thermal fluctuations are expected to play a key role in its allosteric stimulation. We are using standard and accelerated conformational sampling molecular dynamics simulations,
which can provide direct insight into the specific role of thermal fluctuations in the allosteric stimulation of attachment proteins.
Collaborators: Matteo Porotto
Methods/Scientific software
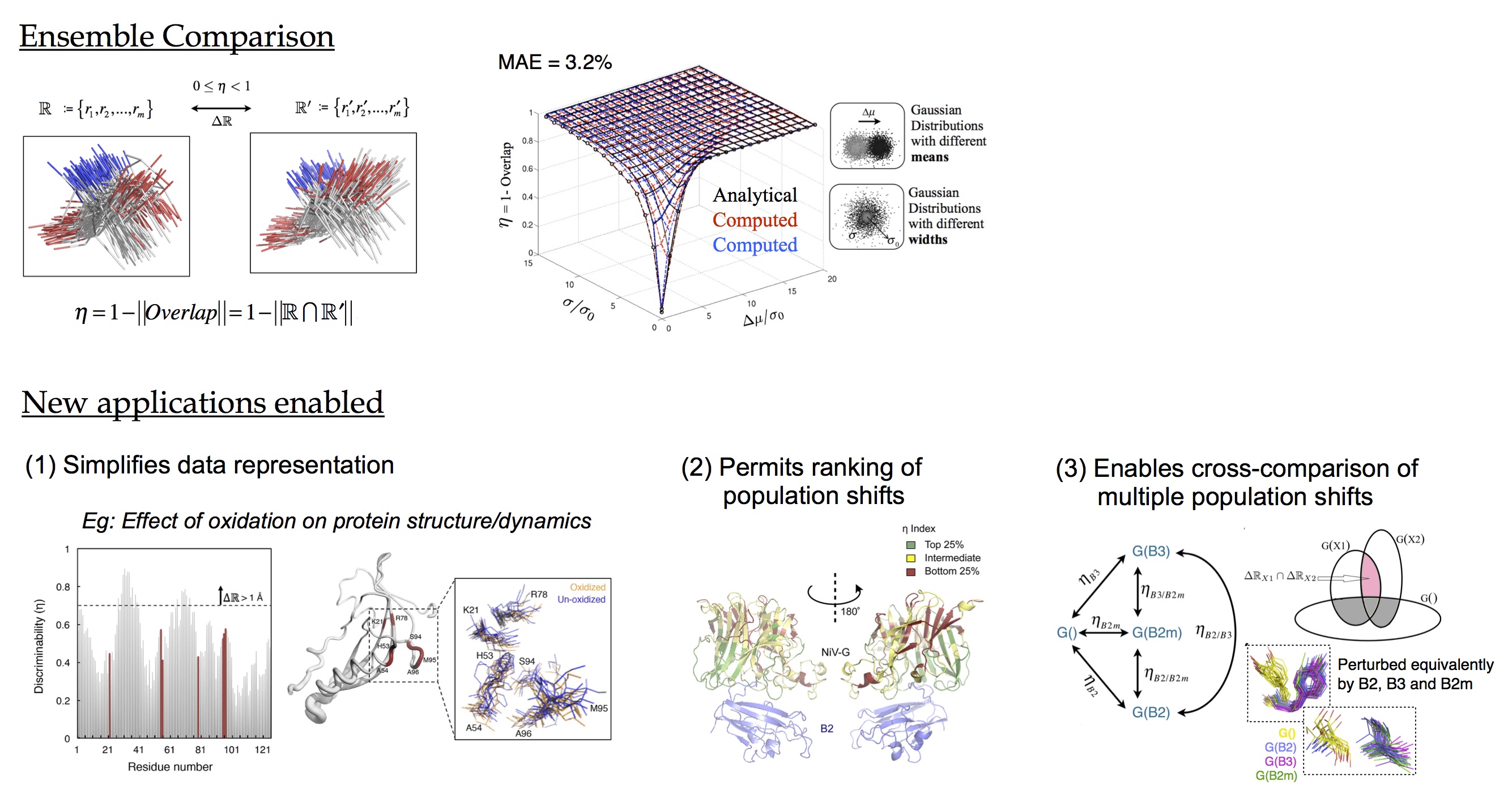
DiCE (Direct Comparison of Ensembles): The traditional approach to compare two or more conformational ensembles is to compare their respective summary statistics, like centers-of-mass (COMs) and root mean square fluctuations (RMSFs). However, if a subset of the summary statistics of the two ensembles is found to be identical, it does not imply that all of the 3n-6 summary statistics of two ensembles will also be identical. The general problem of finding and choosing a feature that appropriately distinguishes two ensembles can be overcome by comparing ensembles directly against each other, and before any dimensionality reduction. A further advantage of comparing ensembles directly against each other is that the resulting quantification naturally embodies differences in conformational fluctuations. We provide the very first method, based on
machine learning, that does just that, and reports the difference between conformational ensembles in terms of a normalized metric relatated directly to the physical overlap between conformation ensembles. The advantage of quantifying differences in terms of a normalized metric is that it allows multi-way comparisons.
Publications: JCTC 2012,
Proteins 2014,
BJ 2016
Source code: MPI-enabled and written using Gromacs APIs. Available at
SimTK.
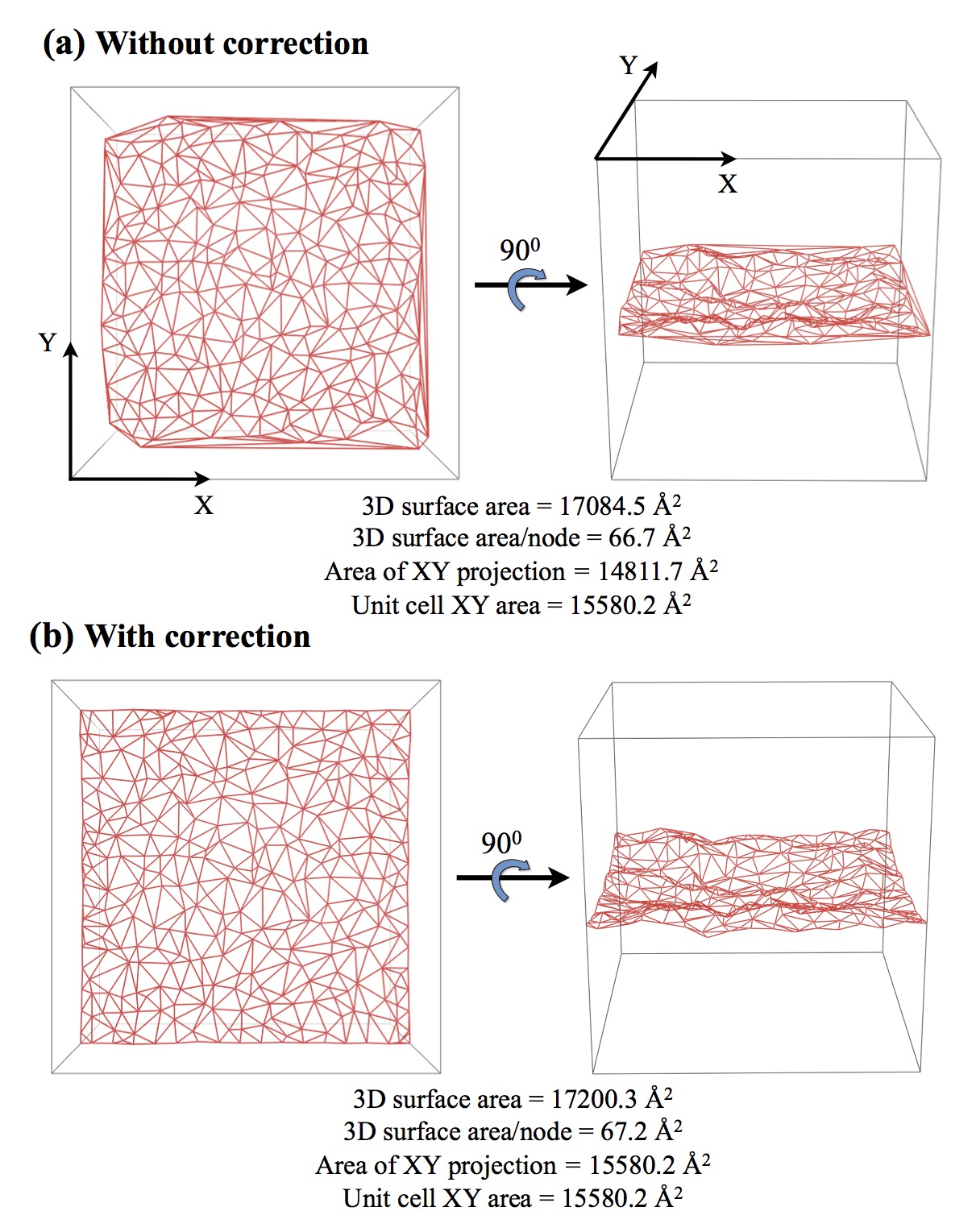
TessLA (Tessellated lipid surface areas): Typically, bilayer areas are determined in one of the two ways: (1) by dividing the partial volume of the hydrocarbon tail by its transverse width; (2) by dividing the lateral area of a leaflet by the number of lipids/leaflet. Both techniques, however, assume that lipid bilayers are geometrically planar. We have developed an algorithm based on Delaunay triangulation to determine the surface area of a non-planar bilayer that accounts for lipid movements across the transverse axis and also corrects for finite size effects.
Publications: Langmuir 2016
Source code: MPI-enabled and written using Gromacs APIs. Available at
SimTK.
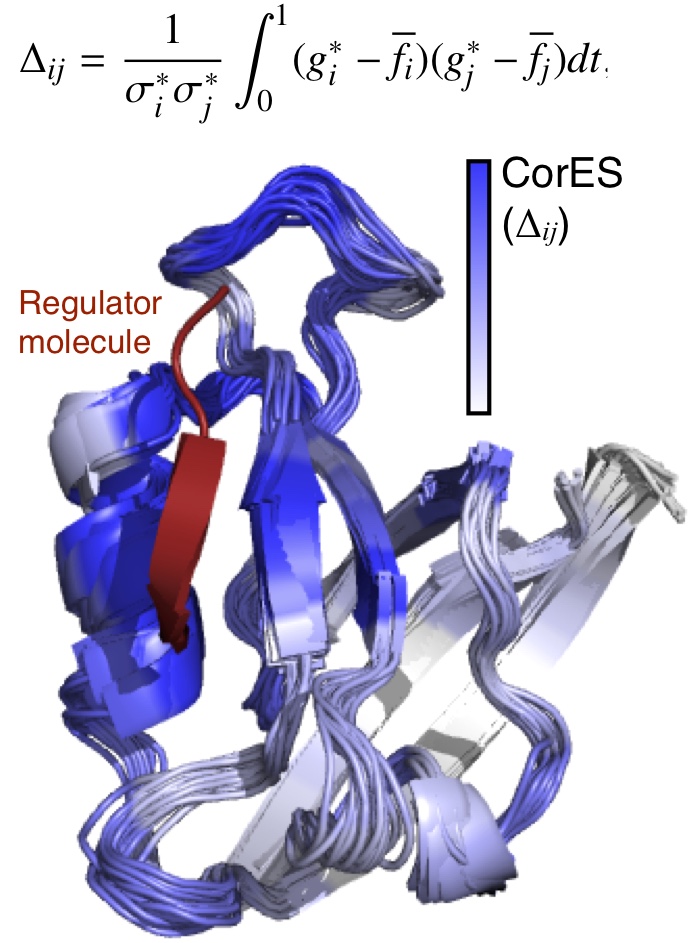
CorES (Correlations in Ensemble Shifts): This method computes the correlation between regulator-induced ensemble shifts at two different sites in a protein. Proteins regulated by dynamic allostery are expected to exhibit a subset of conformational densities that are common to different states, and regulators control protein activity by simply introducing a bias on those densities. A proposition that follows directly from this governing principle is the possibility that perhaps regulator-induced density shifts at two sites in a protein may be correlated. We find this to be true in the case of the PDZ domains, and also that these correlations depends inversely on the distance from the regulator binding site.
Publications: JCP 2018,
Structure 2019
Source code: Under development.